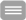
Hereditäre Sensorische und Autonome Neuropathien (HSAN)
Hereditäre Sensorische und Autonome Neuropathien (HSAN)
|
Subtyp |
OMIM-Eintrag |
Gensymbol |
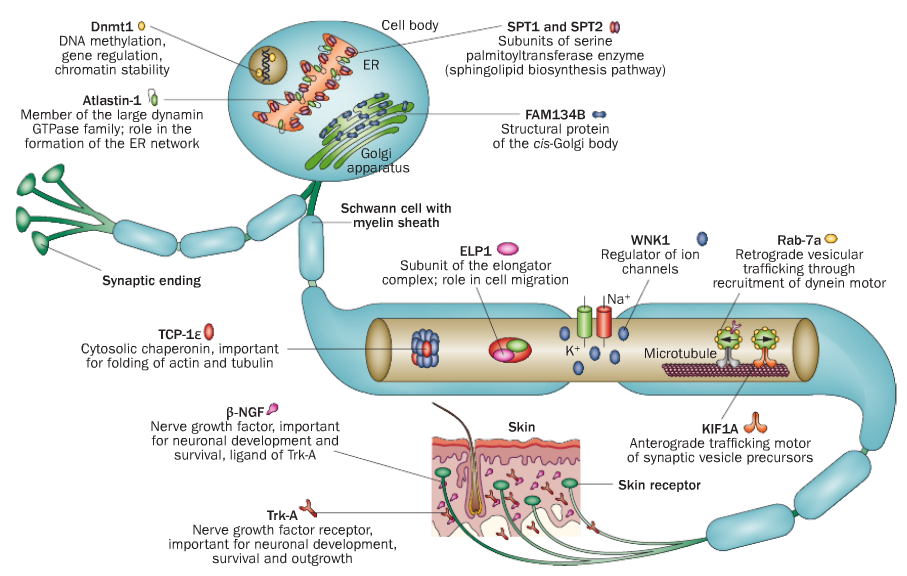
| HSAN1 | OMIM 162400 | SPTLC1 |
| OMIM 613640 | SPTLC2 | |
| OMIM 600882 | RAB7A | |
| OMIM 613708 | ATL1 | |
| OMIM 615632 | ATL3 | |
| OMIM 614116 | DNMT1 | |
| HSAN2 | OMIM 201300 | WNK1/HSN2 |
| OMIM 613115 | FAM134B | |
| OMIM 614213 | KIF1A | |
| OMIM 243000 | SCN9A | |
| HSAN3 | OMIM 223900 | IKBKAP |
| HSAN4 | OMIM 256800 | NTRK1 |
| HSAN5 | OMIM 608654 | NGFB |
| HSAN6 | OMIM 614653 | DST |
| HSAN7 | OMIM 615548 | SCN11A |
| HSAN8 | OMIM 616488 | PRDM12 |
Schmerzassoziierte Erkrankungen (Channelopathies)
|
Subtyp |
OMIM-Eintrag |
Gensymbol |
| Congenital Indifference to Pain (CIP) |
OMIM 243000 | SCN9A |
| Paroxysmal Extreme Pain Disorder (PEPD) |
OMIM 167400 | SCN9A |
| Erythromelalgia, primary (PE) |
OMIM 133020 | SCN9A |
| Small Fiber Neuropathy (SFN) |
OMIM 133020 | SCN9A |
| |
||
| Familial Episodic Pain 2 / Small Fiber Neuropathy | OMIM615551 | SCN10A |
| Congenital Indifference to Pain (CIP) | OMIM 615548 | SCN11A |
| Familial Episodic Pain 3 | OMIM 615552 |
SCN11A |
| Familial Episodic Pain 1 | OMIM 615040 | TRPA1 |
| Methode: |
1) Sanger-Sequenzierung kodierender Exons 2) Panel-Diagnostik, Next-Generation Sequencing, NGS |
| Material: |
DNA-Probe oder EDTA-Blut (3-5 ml) |
| Dauer: |
ca. 8 Wochen |
Pathophysiologie und Genetik:
Die hereditären sensorischen Neuropathien bilden eine klinisch und genetisch heterogene Gruppe seltener Erkrankungen. Bislang wurden mehr als 10 Gene als ursächlich für die HSAN identifiziert. Durch den Verlust von Neuronen, die Schmerz- und Temperaturempfinden, Oberflächen- und Tiefensensibilität sowie Lage- und Tastsinn vermitteln, wird deren zentrale Bedeutung zum Schutz des Körpers offensichtlich. Fehlbelastungen, unbemerkte Verletzungen oder schädigende Temperatureinflüsse führen bei den Betroffenen zu chronifizierenden Ulzerationen mit der Notwendigkeit von Amputationen im Bereich der Extremitäten. Autonome Störungen mit eingeschränkter Herzfrequenzvariabilität, Blutdruck- regulationsstörungen oder gastrointestinale Beschwerden können den Sensibilitätsverlust begleiten. Eine Beteiligung peripherer motorischer Fasern führt zu neurogenen Muskelatrophien mit Kraftverlust. Tendenziell manifestieren sich die autosomal-dominanten Formen der HSAN später als die rezessiven Erkrankungen. Elektrophysiologische Untersuchungen zeigen Zeichen einer axonalen Neuropathie und damit verbundene reduzierte sensible Nervenleitgeschwindigkeiten. Die Nervenbiopsie kann differenzial- diagnostische Hinweise geben.
Diagnostik:
Nach der Isolierung von genomischer DNA aus EDTA-Blut werden nach Absprache die kodierenden Exons der o.g. Gene amplifiziert und mittels Sequenzierung auf Mutationen untersucht.
Wissenschaftliches Interesse an HSAN:
Wir führen bei unklaren HSAN-Fällen bei geeignetem Familienstammbaum genomweite Kopplungsanalysen im Rahmen von wissenschaftlichen Kollaborationen durch. In ausgewählten Fällen können die Proben auch in Absprache mit dem Einsender mittels Next-Generation-Sequencing analysiert werden.
Ansprechpartner:

Arbeitsgruppenleiterin