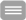
Leistungsspektrum
Stand 05/2023
A
AbortdiagnostikAbortdiagnostik
Für die Untersuchung von Abortmaterial wird die Probe zunächst geteilt: Aus einem Teil der Probe wird DNA isoliert, der andere Teil wird kultiviert und für die Chromosomenanalyse verwendet. Ergibt diese Chromosomenanalyse einen unauffälligen weiblichen Karyotyp, erfolgt eine PCR-basierte Mikrosatellitenanalyse, bei der das Abortmaterial mit dem mütterlichen EDTA-Blut verglichen wird, um eine Kontamination des Untersuchungsmaterials mit mütterlichem Gewebe auszuschließen.
Gelegentlich ist die Kultivierung des Abortmaterials nicht erfolgreich. In diesem Fall kann PCR-basierend eine Untersuchung auf numerische Veränderungen der Chromosomen 13, 15, 16, 18, 21, 22 und XY erfolgen.
| Methode: | Chromosomenanalyse, PCR, Kapillarelektrophorese |
| Material: |
mind. 10 mg Abortmaterial in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) 2 - 5 ml mütterliches EDTA-Blut |
| Dauer: |
ca. 3-4 Wochen |

Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)

Arbeitsgruppenleiterin
Adenomatöse Polyposis coli
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
AFP-Bestimmung
| Methode: | biochemisch |
| Material: |
15 – 20 ml Fruchtwasser in sterilem Probengefäß (nur in Kombination mit einer konventionellen |
| Dauer: | ca. 7 - 10 Tage (Mitteilung mit dem vorläufigen Chromosomenbefund) |
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
Akute Lymphatische Leukämie - ALL (Chromosomenanalyse)
| Methode: | Karyotyperstellung / FISH |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional für FISH: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |

Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Akute Myeloische Leukämie - AML (Chromosomenanalyse)
| Methode: | Karyotyperstellung / FISH |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional für FISH: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Amelogenesis imperfecta
| Methode: |
Next-Generation-Sequenzierung (NGS) |
| Material: | 500 ng DNA oder 2-5 ml EDTA-Blut |
| Dauer: | ca. 8-10 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Amelogenesis imperfecta ist eine erblich bedingte Störung der Zahnschmelzbildung. An diesem Prozess ist eine Vielzahl von Proteinen beteiligt, die bei Mutationen zum klinischen Bild der Amelogenesis imperfecta führen. Abhängig davon, welches Protein durch eine Veränderung in seiner Funktion beeiträchtigt ist, kommt es zur Ausprägung von verschiedenen klinischen Symptomen. Es kann ein autosomal dominanter, autosomal rezessiver oder ein X-chromosomaler Erbgang vorliegen.
Ansprechpartner:
Arbeitsgruppenleiterin
| Methode: | Agilent 180K Array |
| Material: | ≥ 50 ng/µl in ≥ 20 µl DNA oder 2 - 5 ml EDTA-Blut |
| Dauer: | ca. 8 Wochen |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Bei der Array-CGH (Array-based Comparative Genomic Hybridization) handelt es sich um eine Analyse, bei der eine simultane Hybridisierung von fluoreszenzmarkierter Patienten- und Kontroll-DNA auf einem Trägerchip mit DNA-Fragmenten (z.B. Oligonukleotiden) erfolgt. Mittels dieser Methode lassen sich Kopienzahlveränderungen der DNA nachweisen, die aufgrund ihrer Größe in der konventionellen Chromosomenanalyse nicht erkannt werden können. Es kann genomweit ein Verlust von genomischen Bereichen (Deletion) und Hinzugewinn (Duplikation) detektiert werden. Die Veränderungen können in ihrer Größe sehr variabel sein und einige hundert bis mehrere Millionen Basenpaare umfassen. Methodisch bedingt können balancierte Chromosomenveränderungen (z.B. Inversionen) nicht erfasst werden. Häufiger Anforderungsgrund für die Array-CGH Diagnostik sind z.B. Patienten mit mentaler Retardierung und/oder Organfehlbildungen und Dysmorphiezeichen. Bei Kindern mit mentaler Retardierung und unauffälliger konventioneller Chromosomenanalyse findet sich bei etwa 10-20% eine mittels Array-CGH nachweisbare Mikroaberration, die die klinische Symptomatik erklärt.
- Indikationskriterien für die Array-CGH sind eine „Geistige Entwicklungsstörung ungeklärter Ätiologie“ oder autistische Verhaltensweisen (Deutsches Ärzteblatt, Jg. 107, Heft 43, 29.10.2010), im Einzelnen:
- Isolierte Intelligenzminderung (IQ<70)
- Geistige Behinderung in Kombination mit dysmorphologischen Merkmalen unter Beteiligung von 2 oder mehr Systemen
- Entwicklungsstörung des Autismus-Formenkreises oder eine Fehlbildung und schwere Funktionsstörung des Gehirns unbekannter Ursache
- Multiple angeborene Fehlbildungen
- Multiple dysmorphologische Merkmale
In begründeten Fällen ist eine Elternuntersuchung mittels dieser Methode angezeigt, um eine Einschätzung der klinischen Relevanz beim Kind identifizierter Veränderungen zu erlauben.
Aufgrund der hohen Komplexität dieser Methode empfehlen wir vor und nach jeder Untersuchung eine humangenetische Beratung.
Ansprechpartner:
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
Array-CGH Ergebnisverifizierung mittels FISH
| Methode: | Metaphase-FISH (ca. 7000 BAC Sonden genomweit verfügbar) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | ca. 10 Tage |
Anforderungsschein: Herunterladen
Weiterführende Informationen
siehe hier zu array-CGH
und hier zu FISH
Ansprechpartner:

Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
Arrhythmogene Kardiomyopathie
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Azoospermiefaktor
AZF-Deletionen (AZFa, AZFb, AZFc)
| Methode: |
SSP-PCR, Fragmentanalyse |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng DNA |
| Dauer: |
ca. 5-7 Tage |
Weiterführende Informationen:
Etwa 50% aller infertilen Männer weisen eine deutliche Reduktion ihrer Samenzellen auf (Oligozoo- oder Azoospermie), wobei die kausalen Ursachen vielfältig sind. Es wird zwischen obstruktiver und nicht-obstruktiver Azoospermie unterschieden. Eine genetische Erkrankung, die bei Männern zu Unfruchtbarkeit führt, ist die Mukovizidose. Auch durch heterozygote Mutationen im CFTR-Gen kann es zu ein- oder beidseitiger Aplasie der Samenleiter kommen (CUAVD bzw. CBAVD). Bei 10-20% der Patienten mit nicht-obstruktiver Oligozoo- oder Azoospermie ist die Ursache der Infertilität durch das Auftreten von drei verschiedenen Mikrodeletionen auf dem langen Arm des Y-Chromosoms assoziiert (AZFa, AZFb und AZFc). Männer mit einer kompletten AZFa-Deletion weisen keine Keimzellen mehr in Ihrem Hodenepithel auf. Liegt dagegen eine partielle AZFa, AZFb, oder eine komplette AZFc-Deletion vor, können noch reife und befruchtungsfähige Spermien im Hodenepithel vorgefunden werden. Es ist deshalb für den Patienten wichtig zu wissen, ob die Ursache der Fertilitätsstörung eine komplette AZFa oder komplette AZFb/c-Deletion ist. Nur bei partiellen AZFa/b und AZFc Deletionen ist die Gewinnung von reifen Spermien im Hoden-Epithele (TESE) zu erwarten.
weiterführende Literatur:
- Peter H. Vogt: AZF deletions and Y chromosomal haplogroups: history and update based on sequence. Hum Reprod Update. 2005 Jul-Aug;11(4):319-36. Pubmed
- M. Simoni, E. Bakker, C. Krausz: EAA/EMQN best practice guidelines for molecular diagnosis of y-chromosomal microdeletions. State of the art 2004. Int J Androl. 2004 Aug; 27 (4):240-9. Pubmed
Ansprechpartner:
Arbeitsgruppenleiterin
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
C
Chimerismusanalyse nach KnochenmarktransplantationChimerismusanalyse nach Knochenmark-Transplantation
| Methode: | FISH (quantitativ) |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Chorea Huntington
| Methode: | PCR, Short Tandem Repeat Typing |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
| Dauer: |
ca. 5-7 Tage |
Anforderungsschein: Herunterladen
Bitte beachten Sie: Das Konsortium für die molekulargenetische Diagnostik bei Chorea Huntington empfielt die Analyse von zwei unabhängig entnommenen Blutproben.
Weiterführende Informationen
Die Chorea Huntington (Morbus Huntington) ist eine Erkrankung des Nervensystems mit einer geschätzten Prävalenz in Europa von etwa 5 auf 100.000 Einwohnern. Erste Anzeichen der Erkrankung treten meist zwischen dem 35. und 45. Lebensjahr auf. Als Frühsymptome können psychische Veränderungen (u. a. Depressionen, Reizbarkeit, Nervosität, Schlafstörungen) auftreten. Im Weiteren ist die Huntington-Krankheit durch unwillkürliche choreatische Bewegungen gekennzeichnet und führt zu einem vollständigen Verlust von motorischer Kontrolle und intellektuellen Fähigkeiten (Demenz). Dieser Prozess wird durch degenerative Veränderungen in den Stammganglien mit Zelluntergang besonders im Corpus striatum verursacht. Das verantwortliche Gen für die Chorea Huntington ist auf Chromosom 4 (4p16.3) lokalisiert. Im sogenannten Huntingtin-Gen liegt bei den Anlageträgern bzw. den Betroffenen eine Verlängerung einer kleinen Einheit des Erbmaterials, der Basenabfolge CAG vor. Das Trinukleotid CAG kodiert für die Aminosäure Glutamin. Der Einbau einer zu langen Abfolge von Glutaminbausteinen in das Huntingtin-Protein bedingt eine abnorme Proteinfunktion. Die Erkrankung wird autosomal dominant vererbt. Somit besteht für jeden Nachkommen eines Betroffenen bzw. Anlageträgers eine statistische Wahrscheinlichkeit von 50%, das veränderte Gen zu erben.
Ansprechpartner:
Arbeitsgruppenleiterin
chromosomale Umbauten - weitere Abklärung
| Methode: | Metaphase-FISH (mulitcolor-FISH, MCB, (cen) M-FISH) |
|
Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Fluoreszenz in situ Hybridisierung (FISH) ist eine Methode zum Nachweis von Nukleinsäuren (in der Regel DNA) in einzelnen Zellen oder auf Chromosomen. Dabei wird eine künstlich hergestellte Sonde aus Nukleinsäure eingesetzt, die über Basenpaarungen an die nachzuweisende Nukleinsäure bindet (Hybridisierung). Der Nachweis wird direkt in der jeweiligen Struktur (in situ) durchgeführt. (nach http://de.wikipedia.org/wiki/In-situ-Hybridisierung)
Dank der FISH-Technik ist der gleichzeitige Nachweis mehrerer Zielsequenzen im Genom möglich (Vielfarben-FISH-Techniken – siehe hier).
Chromosomale Umbauten können mittels FISH genau analysiert und charakterisiert werden.
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Chromosomenanalyse bei hämatologischen Neoplasien
| Methode: | Karyotyperstellung nach GTG-Bänderung (CBG, NOR) |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Chromosomenanalyse postnatal
| Methode: | Karyotyperstellung |
| Material: |
2 - 5 ml Heparin-Blut Hautfibroblasten / OP-Material ab 15 mg in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) |
| Bearbeitungsdauer: |
OP-Material: ca. 14 - 28 Tage Heparin-Blut: 1-2 Monate (therapierelevante und eilige Fälle bitte kennzeichnen, diese werden innerhalb von 2-3 Wochen bearbeitet) |
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
Chromosomenanalyse pränatal
| Methode: | Karyotyperstellung |
| Material: |
Abortmaterial, Chorion (Placenta): mind. 10 mg in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) Fruchtwasser: 15 - 20 ml in sterilem Probengefäß zum Ausschluss einer mütterlichen Kontamination bei weiblichem Karyotyp bitte 5 ml EDTA-Blut |
| Dauer: |
Abortmaterial: ca. 14 - 28 Tage Chorion/Placenta: Vorläufiger Befund aus der Direktpräparation ca. 1 - 2 Tage, Endbefund ca. 14 - 18 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
Chronische Lymphatische Leukämie - CLL (Chromosomenanalyse)
| Methode: | Karyotyperstellung / FISH (p53-Deletion; 13q14-Deletion; Trisomie 12; ATM/IGH-Genveränderungen) |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Chronische Myeloische Leukämie - CML
| Methode: |
BCR-ABL, qualitativer nachweise (Multiplex-PCR) BCR-ABL, quantitative Bestimmung (Realtime-PCR) BCR-ABL, Mutationsanalyse (Sanger-Sequenzierung) |
| Material: |
EDTA-Blut (20 ml), EDTA-Knochenmark (5 ml) |
| Dauer: | ca. 2 - 14 Tage |

Chronische Myeloische Leukämie - CML (Chromosomenanalyse)
| Methode: | Karyotyperstellung / FISH (Translokation t(9;22)) |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Cowden-Syndrom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Cri-du-Chat-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Katzenschrei-Syndrom oder Cri-du-chat-Syndrom ist erstmals 1963 von dem französischen Genetiker und Kinderarzt Jérôme Lejeune unter wissenschaftlichen Gesichtspunkten beschrieben worden. Er benannte es nach dem katzenähnlichen Schreien (franz.: cri du chat = „Katzenschrei“) der betroffenen Kinder im frühen Kindesalter.
Die Ursache des CDC-Syndroms ist eine strukturelle Chromosomenaberration (nicht numerisch) mit partieller Deletion (= Stückverlust) am kurzen Arm eines Chromosoms 5 (= partielle Monosomie). Der Verlust erfolgt in der Regel zufällig und nach heutigem Wissen ohne besondere äußere Einflüsse im Zeitraum der letzten Zellteilung der Eizelle.
In 15 % der Fälle wird das CDC-Syndrom durch eine unbalancierte Chromosomentranslokation ausgelöst, wobei bei 10 % der Kinder bei einem Elternteil bereits ein Teil des entsprechenden Chromosomenarmes abgebrochen ist und sich an einem anderen Chromosom angelagert hat (= balancierte Translokation). Dieser Elternteil hat kein CDC-Syndrom, da bei ihm die Translokation balanciert (= ausgeglichen) vorliegt und die Menge des Erbmaterials sich somit nicht verändert hat.
Schätzungsweise eines von 50.000 Kindern hat ein CDC-Syndrom, wobei es wahrscheinlich ist, dass das Syndrom oft nicht erkannt bzw. nicht als solches diagnostiziert wird.
Im Verhältnis 5:1 sind deutlich mehr Mädchen als Jungen von dieser Chromosomenbesonderheit betroffen (= Gynäkotropie).
(nach http://de.wikipedia.org/wiki/Katzenschrei-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Cystische Fibrose (CF, Mukoviszidose)
| Methode: |
Untersuchung auf 31 häufigste Mutationen im CFTR-Gen SSP-PCR, Fragmentanalyse (CFTR-Assay Fa. Elucigene) Komplettanalyse CFTR-Gen Next-Generation-Sequenzierung und MLPA |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
| Dauer: |
Screening häufigste Mutationen: 7 - 10 Tage Komplettanalyse: ca. 8 Wochen |
Anforderungsschein: Herunterladen
Unser Labor nimmt regelmäßig an den vom Cystic Fibrosis European Network auf europäischer Ebene durchgeführten Qualitätskontrollen teil.
Weiterführende Informationen:
Die Cystische Fibrose ist eine der häufigsten Stoffwechselerkrankungen in Populationen kaukasischer Abstammung und wird autosomal-rezessiv vererbt. In Deutschland tritt sie mit einer Häufigkeit von etwa 1:3300 Neugeborenen auf. Die Frequenz der heterozygoten Genträger beträgt ca. 1 auf 29. Bei diesem Krankheitsbild handelt sich um eine generalisierte Dysfunktion der exokrinen Drüsen, die im klassischen Fall Atemwege, Verdauungssystem und Reproduktionstrakt betreffen kann. Der Erkrankung liegt eine Funktionsstörung eines Chlorid-Ionenkanals in der apikalen Membran von Drüsenepithelzellen zugrunde, die zur Änderung des Salzgehaltes des Schweißes und anderer Körpersekrete führt. Dadurch kommt es u. a. zur Bildung des charakteristischen, zähflüssigen Schleims, der eine chronische Besiedlung der Lungen mit pathogenen Keimen begünstigt. Auch der Magen-Darm-Trakt kann durch Sekretionsstörungen des Pankreas betroffen sein. Der Übergang zwischen typischer CF, milden und aberranten Formen sowie asymptomatischen Verläufen ist fließend. Aufgrund dieser Variabilität ist es schwer, anhand des Mutationsmusters Vorhersagen über Verlauf und Schwere des Organbefalls zu treffen.
Häufige Gründe für die Anforderung der Diagnostik:
-
Klinischer Verdacht auf Cystische Fibrose (Mekoniumileus, rezidivierende Bronchitiden und Pneumonien, Pankreasinsuffizienz, positiver Schweißtest, hyperechogener fetaler Darm u.a.)
-
positive Familienanamnese für Cystische Fibrose
-
Überprüfung des Heterozygotenstatus bei gesunden Ratsuchenden mit Kinderwunsch
-
obstruktive Azoospermie und Aplasie eines oder beider Samenleiter (CUAVD bzw. CBAVD). Bei etwa 70-80% der Männer mit congenitaler Aplasie des vas deferenz (CAVD) ohne zusätzliche Auffälligkeiten im Urogenitaltrakt werden Mutationen im CFTR-Gen nachgewiesen.
-
Überprüfung des Heterozygotenstatus bei Kinderwunsch vor reproduktionsmedizinischen Verfahren
Weiterführende Links:
Ansprechpartner:
Arbeitsgruppenleiterin
D
DiGeorge- / Velocardiofaciales-Syndrom (CATCH22)DiGeorge- / Velocardiofaciales-Syndrom (CATCH22)
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2-3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das DiGeorge Syndrom beruht auf einer gestörten Entwicklung der dritten und vierten Kiemenbogentasche. Dies führt zu einer Anlage/Entwicklungsstörung einer Reihe von Organen, die aus diesen Strukturen hervorgehen: u.a. Aortenbogen, Thymus, Nebenschilddrüse, Gaumenbogen und Schlundmuskulatur, Gehörgänge und Zahnanlagen. Das klinische Spektrum dieser Störungen ist außerordentlich breit. Dementsprechend ist die Nomenklatur divers und etwas verwirrend. Der Begriff DiGeorge-Syndrom wird für Kinder verwandt, die sich in der Neonatalperiode mit typischem Herzfehler, Thymushypoplasie und Hypocalzämie präsentieren. Der Begriff Velocardiofaciales (Sphrintzen) – Syndrom wird für ältere Kinder verwandt, bei denen die nasale Sprache durch velopharyngeale Inkompetenz im Vordergrund steht. CATCH22 ist ein kollektives Akronym für die unterschiedlich ausgeprägten Syndrome (cardiac abnormality, abnormal facies, T cell deficiency, cleft palate, hypoparathyroidism resulting from 22q11 deletion).
Das Spektrum des Immundefekts durch die Thymushypo- oder aplasie bei DiGeorge-Syndrom ist ebenfalls breit. Es gibt Patienten, die keinerlei T-Zell Abnormalitäten haben, Patienten, die niedrige T-Zell Zahlen haben, aber weitgehend normale T-Zell Funktion (partielles DiGeorge-Syndrom, < 10%) und Patienten, die fast keine T-Zellen haben (komplettes DiGeorge Syndrom, < 1%). Vor allem diese schwer immundefizienten Patienten müssen früh identifiziert werden, um entsprechende Prophylaxe, Therapie und klinisch-immunologische Anbindung zu gewährleisten.
Die meisten Formen des DiGeorge Syndroms sind mit einer Mikrodeletion der Region 22q11.2 verbunden. In seltenen Fällen ist auch eine Assoziation mit einer Hemizygotie für 10p13 beschrieben. Auf molekularer Ebene wurde die Deletion des Transkriptionsfaktors TBX1 als eine wesentliche Determinante für das das klinische Bild des 22q11.2 Deletionssyndroms identifiziert. Molekulargenetische Untersuchungen spielen jedoch für die Diagnostik noch keine Rolle. Es ist wichtig, zu wissen, dass bis zu 5-10% der Patienten mit DiGeorge-Syndrom keine 22q11-Deletion aufweisen.
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Dilatative Kardiomyophatie
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
DNA-Isolierung und Asservierung
| Methode: | Magnetpartikel-assistiert (Qiagen EZ1 Kits, automatisiert) |
| Material: |
2-5 ml EDTA-Blut 15-20 ml Fruchtwasser 10 mg Chorionzotten, Abortmaterial oder Fibroblasten |
| Dauer: |
ca. 7 Tage |
Bitte teilen Sie uns mit, falls die DNA früher als 7 Tage nach dem Eingangsdatum benötigt wird!
Anforderungsschein: Herunterladen
Ansprechpartner:
Arbeitsgruppenleiterin
E
Exomsequenzierung|
Methode: |
Next-Generation-Sequenzierung (NGS) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 3 Monate |
|
untersuchte Gene: |
siehe Liste |
Weiterführende Informationen
Arbeitsgruppenleiterin
F
Familiärer Brust- und EierstockkrebsFamiliärer Brustkrebs (BRCA1/2)
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Brustkrebs (Mammakarzinom) stellt mit einem Anteil von ca. 30% aller Krebserkrankung mit Abstand die häufigste Tumorerkrankung bei Frauen in Deutschland dar, während Eierstockkrebs (Ovarialkarzinom) ca. 3% aller Krebsneuerkrankungen bei Frauen ausmachen. Ungefähr 5 – 10 % aller Mammakarzinome bzw. 10 - 25 % der Ovarialkarzinome sind erblich bedingt.
Folgende Kriterien sind für die molekulargenetische Testung für die bei einer Indexperson in der Familie angesetzt (Deutsches Konsortium Familiärer Brust- und Eierstockkrebs):
- mindestens drei Frauen mit Mammakarzinomerkrankungen unabhängig vom Alter
- mindestens zwei Frauen mit Mammakarzinomerkrankungen, davon eine vor dem 51. Geburtstag
- mindestens eine an Brustkrebs erkrankte Frau und mindestens eine an Eierstockkrebs erkrankte Frau oder eine an Brust- und Eierstockkrebs erkrankte Frau
- mindestens zwei Frauen mit Ovarialkarzinomerkrankungen unabhängig vom Alter
- mindestens eine Frau mit Mammakarzinomerkrankung vor dem 36. Geburtstag
- mindestens eine Frau mit einer bilateralen Mammakarzinomerkrankung, die erste vor dem 51. Geburtstag
- mindestens ein Mann mit Mammakarzinomerkrankung und eine Frau mit Mamma- oder Ovarialkarzinomerkrankung unabhängig vom Alter
- mindestens eine Frau mit triple-negativer Mammakarzinomerkrankung vor dem 60. Geburtstag*
- mindestens eine Frau mit Ovarialkarzinomerkrankung vor dem 80. Geburtstag*
- genetische Veränderung (Mutation) in einem Risikogen für Brust- und/oder Eierstockkrebs in der blutsverwandten Familie
Hauptsächlich prädisponieren pathogene Mutationen im BRCA1- und BRCA2-Gen für das Auftreten von Brust- und Eierstockkrebs. BRCA1-Mutationen sind gegenüber BRCA2-Veränderungen mit einem höheren Lebenszeitrisiko für einen Eierstockkrebs assoziiert. Bei Mutationsträgern besteht ein erhöhtes Risiko für assoziierte Tumoren, wie Darm-, Prostata-, Pankreas-, Hautkrebs und Leukämien. Darüber hinaus sind weitere Gene im Zusammenhang mit der Erkrankung. In Anlehnung der Empfehlung des deutschen Konsortiums Familiärer Brust- und Eierstockrebs werden Erfüllung der o.g. Kritierien bei uns von 13 Gene analysiert: BRCA1, BRCA2, ATM, BARD1, BRIP1, CDH1, CHEK2, PALB2, PTEN, RAD51C, RAD51D, STK11 und TP53.
Ansprechpartner:
Arbeitsgruppenleiterin
Familiäres Melanom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
H
Hereditäre Sensomotorische Neuropathien (HMSN)Hereditäre Sensomotorische Neuropathien (HMSN)
| Subtyp | Gensymbol | OMIM-Eintrag |
| CMT1A/HNPP | PMP22*,° | |
| CMT1B | MPZ° | OMIM 118200 |
| CMTX1 | GJB1° | OMIM 302800 |
| CMT2A2 | MFN2° | OMIM 609260 |
*Duplikations-/Deletionsanalyse mittels MLPA-Kit
°Direktsequenzierung
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Hintergrund
Es handelt sich bei der HMSN um eine genetisch sehr heterogene, erbliche Erkrankung des Nervensystems, die sowohl die motorischen Fähigkeiten (Bewegung) als auch die sensorischen Fähigkeiten (Empfindung) beeinflussen kann. Die Erkrankung wird nach Ihren Entdeckern auch Charcot-Marie-Tooth (CMT) genannt. Die HMSN ist eine Erkrankung der peripheren Nerven, bei der die isolierende Schicht um die Nerven (Myelinschicht) und/oder die Nervenfasern selbst zugrunde gehen. Charakteristisch ist eine symmetrische, distal betonte Muskelschwäche bzw. - atrophie. Meistens sind zunächst die Fuß- und Wadenmuskulatur betroffen, später auch die Handmuskeln. Fußdeformitäten wie Hohlfüße und Krallenzehen gehören zum klinischen Bild. Die sensiblen Ausfälle sind im Allgemeinen geringer ausgeprägt. In den meisten Fällen ist die Erkrankung langsam progredient, mit sehr variablem Verlauf. In den meisten Fällen folgt die HMSN einem autosomal dominanten Erbgang. Dies gilt sowohl für die HMSN I (demyelinisierender Typ) als auch für die HMSN II (axonaler Typ). Die X-chromosomale Vererbung ist bei der HMSN ebenfalls nicht selten. Insgesamt liegt molekulargenetisch am Häufigsten eine Duplikation des PMP22-Gens zugrunde. Die entsprechende Deletion des PMP22-Gens führt zu einer Neuropathie mit Neigung zu Druckläsionen.
Vorgeschlagenes diagnostisches Vorgehen aus:
Szigeti K, Lupski JR., Eur J Hum Genet. 2009 Jun;17(6):703-10
Ansprechpartner:
Arbeitsgruppenleiterin
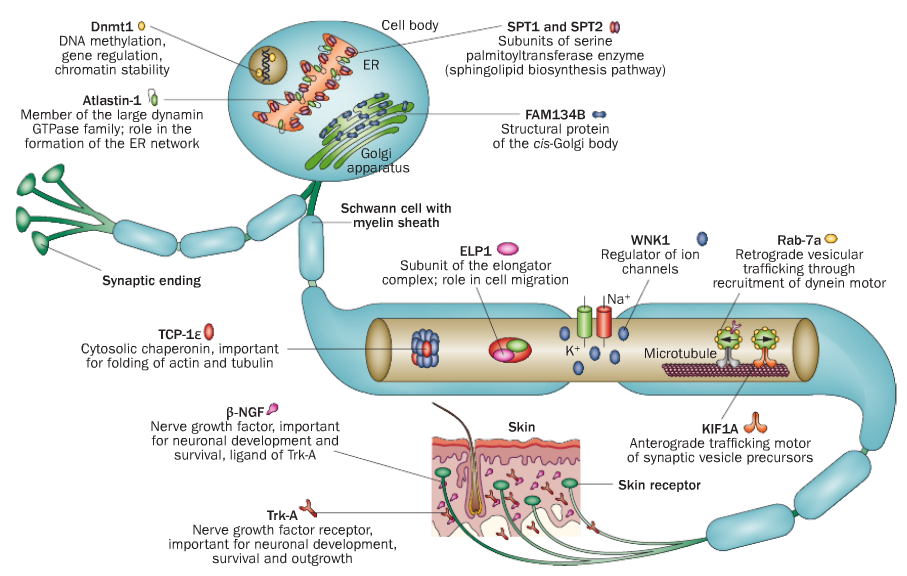
Hereditäre Sensorische und Autonome Neuropathien (HSAN)
|
Subtyp |
OMIM-Eintrag |
Gensymbol |
| HSAN1 | OMIM 162400 | SPTLC1 |
| OMIM 613640 | SPTLC2 | |
| OMIM 600882 | RAB7A | |
| OMIM 613708 | ATL1 | |
| OMIM 615632 | ATL3 | |
| OMIM 614116 | DNMT1 | |
| HSAN2 | OMIM 201300 | WNK1/HSN2 |
| OMIM 613115 | FAM134B | |
| OMIM 614213 | KIF1A | |
| OMIM 243000 | SCN9A | |
| HSAN3 | OMIM 223900 | IKBKAP |
| HSAN4 | OMIM 256800 | NTRK1 |
| HSAN5 | OMIM 608654 | NGFB |
| HSAN6 | OMIM 614653 | DST |
| HSAN7 | OMIM 615548 | SCN11A |
| HSAN8 | OMIM 616488 | PRDM12 |
Schmerzassoziierte Erkrankungen (Channelopathies)
|
Subtyp |
OMIM-Eintrag |
Gensymbol |
| Congenital Indifference to Pain (CIP) |
OMIM 243000 | SCN9A |
| Paroxysmal Extreme Pain Disorder (PEPD) |
OMIM 167400 | SCN9A |
| Erythromelalgia, primary (PE) |
OMIM 133020 | SCN9A |
| Small Fiber Neuropathy (SFN) |
OMIM 133020 | SCN9A |
| |
||
| Familial Episodic Pain 2 / Small Fiber Neuropathy | OMIM615551 | SCN10A |
| Congenital Indifference to Pain (CIP) | OMIM 615548 | SCN11A |
| Familial Episodic Pain 3 | OMIM 615552 |
SCN11A |
| Familial Episodic Pain 1 | OMIM 615040 | TRPA1 |
| Methode: |
1) Sanger-Sequenzierung kodierender Exons 2) Panel-Diagnostik, Next-Generation Sequencing, NGS |
| Material: |
DNA-Probe oder EDTA-Blut (3-5 ml) |
| Dauer: |
ca. 8 Wochen |
Pathophysiologie und Genetik:
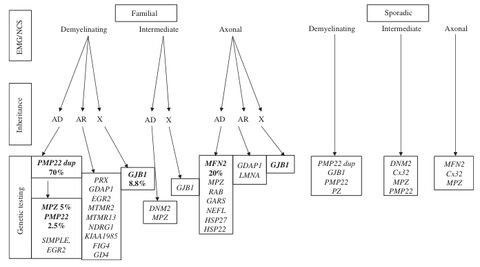
Die hereditären sensorischen Neuropathien bilden eine klinisch und genetisch heterogene Gruppe seltener Erkrankungen. Bislang wurden mehr als 10 Gene als ursächlich für die HSAN identifiziert. Durch den Verlust von Neuronen, die Schmerz- und Temperaturempfinden, Oberflächen- und Tiefensensibilität sowie Lage- und Tastsinn vermitteln, wird deren zentrale Bedeutung zum Schutz des Körpers offensichtlich. Fehlbelastungen, unbemerkte Verletzungen oder schädigende Temperatureinflüsse führen bei den Betroffenen zu chronifizierenden Ulzerationen mit der Notwendigkeit von Amputationen im Bereich der Extremitäten. Autonome Störungen mit eingeschränkter Herzfrequenzvariabilität, Blutdruck- regulationsstörungen oder gastrointestinale Beschwerden können den Sensibilitätsverlust begleiten. Eine Beteiligung peripherer motorischer Fasern führt zu neurogenen Muskelatrophien mit Kraftverlust. Tendenziell manifestieren sich die autosomal-dominanten Formen der HSAN später als die rezessiven Erkrankungen. Elektrophysiologische Untersuchungen zeigen Zeichen einer axonalen Neuropathie und damit verbundene reduzierte sensible Nervenleitgeschwindigkeiten. Die Nervenbiopsie kann differenzial- diagnostische Hinweise geben.
Diagnostik:
Nach der Isolierung von genomischer DNA aus EDTA-Blut werden nach Absprache die kodierenden Exons der o.g. Gene amplifiziert und mittels Sequenzierung auf Mutationen untersucht.
Wissenschaftliches Interesse an HSAN:
Wir führen bei unklaren HSAN-Fällen bei geeignetem Familienstammbaum genomweite Kopplungsanalysen im Rahmen von wissenschaftlichen Kollaborationen durch. In ausgewählten Fällen können die Proben auch in Absprache mit dem Einsender mittels Next-Generation-Sequencing analysiert werden.
Ansprechpartner:
Arbeitsgruppenleiterin
Hereditäre Spastische Paraplegie (HSP)
| Subtyp | Gensymbol | OMIM-Eintrag |
| SPG3A | ATL1 | OMIM 182600 |
| SPG4 | SPAST | OMIM 182601 |
| SPG31 | REEP1 | OMIM 610250 |
| Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
| Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Pathophysiologie und Genetik:
Die Hereditären Spastischen Paraplegien (HSP) bilden eine klinisch und genetisch heterogene Gruppe von Erkrankungen. Sie kann einem autosomal dominanten (in 70-80%), autosomal rezessiven (in ca. 20%) oder einem X-chromosomalen (selten) Erbgang folgen. Unter den häufigen autosomal-dominanten Formen finden sich am Häufigsten Mutationen in den im Rahmen der Diagnostik angebotenen Genen ATL1, SPAST und REEP1. Es werden reine/unkomplizierte und komplizierte Formen der Erkrankung unterschieden. Zu den Hauptsymptomen der reinen Form gehören eine spastische Tonuserhöhung der Muskulatur, insbesondere der unteren Extremitäten, gesteigerte Muskeleigenreflexe, eine Muskelschwäche, reduziertes Vibrationsempfinden sowie Blasenentleerungsstörungen. Bei den komplizierten Formen können eine mentale Retardierung, Epilepsie, Demenz, Ataxie, Taubheit und Optikusatrophie hinzukommen. Das Institut beschäftigt sich auch wissenschaftlich mit der HSP (Funktionelle Genetik).
Links: Tom Wahlig Stiftung
Ansprechpartner:
Arbeitsgruppenleiterin
Hypertrophe Kardiomyophatie
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Die hypertrophe Kardiomyopathie (HCM) ist eine autosomal-dominant vererbte strukturelle Erkrankung des Herzens, die zu einer Verdickung des Herzmuskels und dadurch charakteristischen Veränderungen im EKG kommt. Häufige Symptome sind Kurzatmigkeit, Atemnot, Schwindel und Ohnmacht und können von mild bis schwer variieren. In ca. 90% der Fälle werden krankheitsursächliche Mutationen in den Genen MYH7, MYBPC3, TNNT2 und TNNI3 identifiziert. Die Hypertrophe Kardiomyopathie geht mit einer Veränderung der Sarkomerproteine einher, wobei ca. 50% der Erkrankungen familiär bedingt sind und sich durch einen variablen Phänotyp auszeichnen (Prävalenz ca. 1:500).
Ansprechpartner:
Arbeitsgruppenleiterin
K
Kallmann-SyndromKallmann-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Als Kallmann-Syndrom (KS) oder olfaktogenitales Syndrom bezeichnet man einen angeborenen Symptomkomplex aus Hypo- bzw. Anosmie (verminderter bis fehlender Geruchssinn) in Verbindung mit Hoden- bzw. Ovarialhypoplasie (Unterfunktion) bedingt durch einen hypogonadotropen Hypogonadismus. Letztere treten jedoch nur in ca. 30 % der bekannten Fälle auf. Benannt ist es nach dem Psychiater Franz Josef Kallmann. Die erste Erwähnung fand es 1856 in einer Veröffentlichung von Aureliano Maestre de San Juan (1828–1890). In spanischsprachigen Teilen der Welt ist das Syndrom immer noch nach diesem benannt (Síndrome de Maestre-Kallman-Morsier). Das Kallmann-Syndrom ist bei Männern (1:10.000) häufiger als bei Frauen (1:50.000). Das Kallmann-Syndrom (KS) selbst ist eine Unterform des Hypogonadismus.
Ursächlich für das Kallmann-Syndrom ist eine Mutation, die eines der Proteine betrifft, die bei der Entwicklung des Bulbus olfactorius und bestimmter Kerngebiete des Hypothalamus eine entscheidende Rolle spielen. Beim KS kommt es genetisch bedingt zu einer Aplasie des Bulbus olfactorius.
Für vier Formen des KS sind die ursächlichen Mutationen bekannt: KAL1 (X-chromosomal rezessiv, Mutation des Proteins Anosmin), KAl2 bis KAL4. Hier angeboten wird die Analyse af eine Mikrodeletion der KAL1-Region.
Ein wichtiger Hinweis auf das KS ist die Hyp- bis Anosmie (stark verminderter bis fehlender Geruchssinn), die teilweise mit einem hypothalamischen Hypogonadismus vergesellschaftet ist. Oft ist dies das einzige beobachtbare Symptom. Bei Männern manifestiert sich das Syndrom mit ausbleibender oder verzögerter Pubertät und fehlender Entwicklung der sekundären Geschlechtsmerkmale (Stimmbruch, Körperbehaarung, Bartwuchs), bei Frauen liegt ebenfalls fehlende Entwicklung der sekundären Geschlechtsmerkmale und Amenorrhoe vor. Die Spiegel von LH und FSH sind vermindert oder auf niedrig-normalem Niveau. Spiegel der Geschlechtshormone sind auf vorpubertärem Niveau. (nach http://de.wikipedia.org/wiki/Kallmann-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Kontaminationsausschluss
Unter verschiedenen Voraussetzungen kann es erforderlich sein, eine Kontamination des (pränatalen) Untersuchungsmaterials mit mütterlichem Gewebe auszuschließen:
- mütterlicher und kindlicher Genotyp sind identisch
- Untersuchungsmaterial ist stark blutig
Dazu ist zwingend EDTA-Blut der Mutter notwendig!
|
Methode: |
PCR, Mikrosatellitenanalyse |
| Material: |
2 - 5 ml mütterliches EDTA-Blut |
| Dauer: |
ca. 1 Woche |
Arbeitsgruppenleiterin
L
Li-Fraumeni-SyndromLi-Fraumeni-Syndrom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Lynch-Syndrom/ HNPCC
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
M
Marfan-SyndromMarfan-Syndrom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Markerchromosomen - Charakterisierung
| Methode: | Metaphase-FISH (multicolor-FISH, MCB, (cen-)M-FISH) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Markerchromosomen / Translokationen Identifizierung
| Methode: | Einfärben- bis 24-Farben-FISH, Multicolor-Bänderung (MCB), (qualitativ) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Mentale Retardierung / Autismus
|
Methode: |
Agilent 180K Array |
|
Material: |
≥ 50 ng/µl in ≥ 20 µl DNA oder 2 - 5 ml EDTA-Blut |
|
Dauer: |
ca. 8 Wochen |
Arbeitsgruppenleiterin
Hinweise:
Der Berufsverband Medizinische Genetik e. V. hat folgende Leitlinie verabschiedet:
Jede molekulargenetische Diagnostik sollte im Rahmen einer genetischen Beratung erfolgen. Dies gilt insbesondere im Hinblick auf Befundmitteilung und -interpretation gegenüber Ratsuchenden.
Mikrodeletionssyndrom 1p36
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Die Monosomie 1p36 ist in den letzten Jahren zunehmend als distinktes Chromosomen-Deletions-Syndrom erkannt worden. Es wird für eines der häufigsten Deletions-Syndrome gehalten. Die Inzidenz bei Geburt ist etwa 1-2 / 10.000. Die Bruchpunkte sind variabel und liegen zwischen den Banden 1p36.13 und 1p36.33. Die Mehrheit der Kinder mit der Deletion 1p36 ist in der Entwicklung retardiert, und dies meist auch schwer, mit Verhaltensanomalien und Autoaggression. Hypotonie und Fütterprobleme mit oropharyngealer Dysphasie sind häufig. Auch zerebrale Krampfanfälle sind häufig und beeinträchtigen die Patienten erheblich. Schließlich gehören noch Dysmorphien, Herzfehler und Seh- und Hörstörungen zum Krankheitsbild. Zur Betreuung gehört auch eine umfassende Beurteilung hinsichtlich der hauptsächlichen klinischen Komplikationen mittels Echokardiographie und ophthalmologischer Untersuchung (nach http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=1606).
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Neuere Erkenntnisse ergaben, dass eine große Zahl klinischer Syndrome aus sog. Mikrodeletionen oder Mikroduplikationen resultiert. Zugrunde liegen bei all diesen klinisch durchaus unterschiedlichen Syndromen molekular das gleiche Prinzip: 1-5 Megabasenpaare sind nur in einer Kopie oder in drei Kopien pro Genom vorhanden. Diese Kopiezahlveränderungen führen aber zu keiner sichtbaren Alteration des Karyotyps, obwohl relativ große Mengen an DNA (Millionen von Basenpaaren) in einem Genom zu wenig bzw. zu viel vorliegen.
Im Folgenden sind Mikrodeletions- und Mikroduplikationssyndrome genannt für die uns Sonden vorliegen – aufgrund der großen Zahl dieser Syndrome können wir hier keine weiterführenden Informationen zu jedem einzelnen Krankheitsbild anbieten.
Mikrodeletionssyndrome / Mikroduplikationssyndrome Liste
Dauer: 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal)
|
Zytogenet. |
Mikro- |
Mikro- |
Referenzen D = Decipher S = Slavotinek, 2008 LL = Lee/Lupski 2006 W = Wessex collection |
| 1pter-p36.31 |
Microdel. |
- |
D |
| 1p34.1 |
- |
Microdup. |
Hanemaaijer, 2009 |
| 1p32.2-p32.3 |
Microdel. |
- |
Mulatinho, 2008 |
| 1q21.1 |
Microdel. |
Microdup. |
D |
| 1q21.1 |
Thrombocytopenia- |
- |
D |
| 1q32.2-q41 |
Van der Waude- |
- |
Wong, 2001 |
| 1q41-q42 |
Microdel. |
- |
S |
| 1q44 |
Corpus callosum agenesie Microdel. Syndrom |
- | van Bon, 2008 |
| 2p24.3 |
Feingold |
- |
W |
| 2p21 |
Hypotonia-Cystinurea- |
- |
W |
| 2p21 |
Holoprosencephaly |
- |
W |
| 2p15-16.1 |
Microdeletion |
- |
D |
| 2q11.2 |
Microdel. |
- |
Steichen- Gersdorf, 2008 |
| 2q13 |
Nephronophthisis |
- |
W |
| 2q22.3 |
Mowat-Wilson |
- |
W |
| 2q23.1 |
Microdel. |
- |
Jaillord, 2008 |
| 2q23.3-q24.1 |
Microdel. |
- |
Lybaek, 2009 |
| 2q31.1 |
Synpolydactyly 1 |
- |
W |
| 2q33.1 |
Microdel. |
- |
D |
| 2q37 |
Microdel. |
- |
D |
| 3p25-p26 |
Distal 3p |
- |
W |
| 3p25-p26 |
Von Hippel |
- |
W |
| 3p21.31 |
Microdel. |
- |
Haldeman- Englert, 2008 |
| 3p14.1-p13 |
Microdel. |
- |
Pariani, 2009 |
| 3q13.11-q13.12 |
Proximal 3q |
- |
Simovich, 2008 |
| 3q23 |
Blepharophimosis, |
- |
Chandler, 1997 |
| 3q24 |
Dandy-Walker- |
- |
W |
| 3q27.3-q29 |
Microdel. |
- |
Pallazzon, 2009 |
| 3q29 |
Microdel. |
Microdup. |
D |
| 4pter-p16.3 |
Wolf-Hirschhorn-Syndrom (WHS) |
- |
D |
| 4q21.1-q21.3 |
Microdel. |
- |
W |
| 4q22.1 |
Parkinson |
- |
LL |
| 4q25 |
Rieger |
- |
W |
|
5p15.2-p15.33 |
Cri-du-Chat- |
- |
D |
| 5p13.2 |
Cornelia de Lange |
- |
W |
| 5q13.2 |
Spinal muscular |
- |
LL |
| 5q14.3 |
Microdel. |
- |
Le Meuer, 2009 |
| 5q14.3-q15 |
Microdel. |
|
Engels, 2009 |
| 5q22.2 |
Familial |
- |
D |
| 5q23.2 |
Adult-onset |
- |
D |
| 5q35.1 |
- |
Pseudotrisomy 13 |
Koolen, 2006 |
| 5q35.1 |
Microdel. |
- |
Baekvad- Hansen, 2006 |
| 5q35.2 |
Parietal Foramina |
- |
Wilkie, 2000 |
| 5q35.2-q35.3 |
Sotos- |
Microdup. |
D/ Kirchhoff 2007 |
| 6p25 |
Microdel. |
- |
D |
| 6p21.3 |
Renal |
- |
W |
| 6q16.2 |
Prader-Willi like |
- |
Faivre, 2002 |
| 6q24.2 |
- |
Microdup. |
Mackay, 2005 |
| 6q25.2-q25.3 |
Microdel. |
- |
Nagamani, 2008 |
| 7p21.1 |
Saethre-Chotzen- |
- |
Johnson, 1998 |
| 7p14.1 |
Greig Cephalo-polysyndactyly |
- |
Johnston, 2003 |
| 7q11.23 |
Williams-Beuren- |
Microdup. |
D |
| 7q21.3 |
Split hand/foot |
- |
D |
| 7q31 |
Speech-Language- |
- |
D |
| 7q36 |
Holoprosencephaly |
- |
W |
| 7q36 |
Currarino |
- |
W |
| 7q36.3 |
- |
Triphalangeal Thumb Polysyndactyly Syndrom |
Klopocki, 2008 |
| 8p23.1 |
Microdel. |
- |
D |
| 8p12p21 |
Microdel. |
- |
Willemsen, 2009 |
| 8q12.2 |
CHARGE |
Microdup. |
Vissers, 2004/ Lehman 2009 |
| 8q21.3-q22.1 |
Microdel. |
- |
S |
| 8q24.11 |
Langer-Giedion- |
- |
Feely, 2002 |
| 9pter-p22.3 |
Monosomy 9p |
- |
W |
| 9p24.3 |
Sex Reversal |
- |
Muroya, 2000 |
| 9q22.3 |
Microdel. |
- |
S |
| 9q22.32 |
Holoprosen- |
- |
W |
| 9q33.3 |
Nail-Patella |
- |
Bongers, 2008 |
| 9q34.3 |
Subtelomere |
- |
D |
| 10p15 |
Hypoparathyroidism, |
- |
van Esch, 2000 |
| 10p12.31 |
Di George |
- |
W |
| 10q22-q23 |
Microdel. |
- |
W |
| 10q23.2-q23.3 |
Juvenile Polyposis |
- |
W |
| 10q24.32 |
- |
Split-Hand/ |
W |
| 11p15.5 |
Beckwith- |
BWS/ SRS |
Russo, 2006 |
| 11p13 |
WAGR- |
- |
D |
| 11p11.2 |
Potocki-Shaffer- |
- |
D |
| 11q14.1-q14.2 |
Microdel. |
- |
W |
| 11q23.3-qter |
Jacobsen |
- |
W |
| 12q14 |
Microdeletion |
- |
D |
| 12q24.1 |
Noonan |
- |
W |
| 13q12.12 |
Spastic ataxia |
- |
Breckpot, 2008 |
| 13q12.3-q13.1 |
Microdel. |
- |
W |
| 13q14.2 |
Retinoblastoma |
- |
W |
| 13q22 |
Hirschsprung Disease |
- |
W |
| 13q32.3 |
Holoprosencephaly5 |
- |
W |
| 14q12 |
Congentital Rett |
Microdup. |
Mencarelli, 2009/ Yeung 2009 |
| 14q22-q23 |
Microdel. |
- |
W |
| 15q11.2-q13.1 |
Angelman-Syndrom |
Microdup. |
D/LL |
| 15q11.2-q13.1 |
Angelman-Syndrom |
Microdup. |
D/LL |
| 15q11.2-q13.1 |
Prader-Willi- |
Microdup. |
D/LL |
| 15q11.2-q13.1 |
Prader-Willi- |
Microdup. |
D/LL |
| 15q13.3 |
Microdel. |
Microdup. |
D/ Miller, 2008 |
| 15q15.3 |
Deafness and Male |
- |
Zhang, 2007 |
| 15q21 |
Microdel. |
- |
Pramparo, 2005 |
| 15q24 |
Microdel. |
Microdup. |
D/ Kiholm Lund, 2008 |
| 15q26.2 |
Fryns |
- |
Slavotinek, 2005 |
| 16p13.3 |
ATR-16- |
- |
D |
| 16p13.3 |
Tuberous Sclerosis |
Tuberous Sclerosis |
Kozlowski, 2007 |
| 16p13.1 |
Microdel. |
Microdup |
Ullmann, 2007 |
| 16p13.3 |
Rubinstein-Taybi- |
- |
D |
| 16p11.2-p12.2 |
Microdel. |
- |
D |
| 16p11.2 |
Microdel. |
- |
Kumar, 2008 |
| 16q11.2-q12.2 |
Microdel. |
- |
Ballif, 2008 |
| 16q21-q22 |
Microdel. |
- |
W |
| 16q24.1 |
Microdel. |
- |
Stankiewicz, 2009 |
| 16q24.3 |
Fanconi Anämie A (FANCA) Del. Syndrom |
- |
W |
| 17p13.3 |
Miller-Diecker- |
MDS |
D/ Roos, 2009 |
| 17p12 |
Hereditary liability to |
Charcot-Marie- |
D |
| 17p11.2 |
Smith-Magenis- |
Potocki- |
D |
| 17q11 |
Neurofibromatose |
Microdup. |
D/ Grisart, 2008 |
| 17q11.2-q12 |
Microdel. |
- |
S |
| 17q12 |
Microdel. |
- |
D |
| 17q12 |
Microdel. |
Microdup. |
S/ Mefford, 2007 |
| 17q12-q21 |
Van Buchem |
- |
W |
| 17q21.3 |
Microdel. |
- |
D |
| 17q22-23.2 |
Microdel. |
- |
Puusepp, 2008 |
| 17q24.2-q24.3 |
Carney Complex |
- |
Blyth, 2008 |
| 18p11.31 |
Holoprosencephaly |
- |
W |
| 18q12.3-q21.1 |
Proximal 18q |
- |
Cody, 2007 |
| 19q13.11 |
Microdel. |
- |
Malan, 2009 |
| 19q13.2 |
Diamond-Blackfan |
- |
Tentler, 2000 |
| 20p12.3 |
Wolff-Parkinson- |
- |
Lalani, 2008 |
| 20p11-p12 |
Alagille- |
- |
Yuan, 1998 |
| 20q13.13-q13.2 |
Microdel. |
- |
S |
| 20q13.32 |
Albright Hereditary |
- |
Aldred, 2002 |
| 21q21.3 |
- |
Microdup. |
D |
| 21q22.12 |
Thrombo- |
- |
Shinawi, 2008 |
| 22p11.1-q11.21 |
- |
Cat Eye- |
D |
| 22q11.21-q11.23 |
Di George Syndrom/ |
Microdup. |
D |
| 22q11.2 |
Distal Microdel. |
Distal Microdup. |
D/ Coppinger, 2009 |
| 22q12.2 |
NF2 Microdel. |
- |
W |
| 22q13 |
Microdel. |
- |
D |
| Xp22.33 |
Leri-Weill |
- |
D |
| Xp22.32-p22.31 |
X-Linked |
- |
W |
| Xp22.31 |
Steroid sulphatase |
- |
D |
| Xp22.32 |
Kallmann- |
- |
Massin, 2003 |
| Xp22 |
MIDAS |
- |
Wimplinger, 2007 |
| Xp21.3-p21.2 |
X-linked congenital |
- |
Muscatelli, 1994 |
| Xp21.2 |
Muscular Dystrophy |
- |
Zneimer, 1993 |
| Xp21.2 |
Complex Glycerol |
- |
Stanczak, 2007 |
| Xp11.3 |
Xp11.3 Del. |
- |
Lugtenberg, 2006 |
| Xp11.23 |
Goltz- |
- |
Grzeschik, 2007 |
| Xp11.22 |
- |
17-beta-hydroxysteroid dehydrogenase X (HSD) |
Froyen, 2008 |
| Xq13.2 |
X-inactivation |
- |
W |
| Xq22.1 |
Bruton Agamma- |
- |
Richter, 2001 |
| Xq22.2 |
Microdel. |
Pelizaeus-Merzbacher |
LL/D |
| Xq22.3-q23 |
Microdel. |
- |
Hoischen, 2009 |
| Xq25 |
Lymphoproliferative |
- |
W |
| Xq27.1 |
- |
X-linked |
Solomon, 2002 |
| Xq27.3 |
Fragile Site Mental |
- |
Coffee, 2008 |
| Xq28 |
Rett- |
MECP2 |
D |
| Xq28 |
early infantile epileptic |
- |
Saitsu, 2008 |
| Yp11.31 |
Sex-Determining |
- |
W |
| Yq11.21 |
AZFa |
- |
D |
| Yq11.221-q11.223 |
AZFb |
- |
D |
| Yq11.221-q11.23 |
AZFb+c |
- |
D |
| Yq11.223-q11.23 |
AZFc |
- |
D |
Dauer: 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal)
Weiterführende Informationen
siehe oben auf dieser Seite
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Miller-Dieker-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Beim Miller-Dieker-Syndrom auch Miller-Dieker-Lissenzephalie oder 17-p-Syndrom handelt es sich um ein Krankheitsbild als Folge einer Chromosomenanomalie im Chromosom 17. In der Folge kommt es zu einer schweren Fehlentwicklung des Großhirns in der Embryonalphase. Das Miller-Dieker-Syndrom geht häufig mit weiteren Entwicklungsfehlern einher. Die Erstbeschreibung erfolgte 1963 durch J. Q. Miller sowie 1969 durch H. Dieker.
Ursache für die Entstehung eines Miller-Dieker-Syndroms ist eine Chromosomenaberration mit Mikrodeletionen im terminalen kurzen Arm von Chromosom 17 im Genlocus p13.3. Hierdurch kommt es in der embryonalen Gehirnentwicklung zu einer Migrationsstörung der Nervenzellen der Großhirnrinde. Die ab der 22. Woche der Embryonalentwicklung stattfindenden Schritte in der Gehirnentwicklung, bei der sich die Hirnfurchen ausbilden bleiben teilweise oder ganz aus. Kinder, die mit dem Miller-Dieker-Syndrom geboren werden weisen eine schwere geistige Behinderung auf. Typisch ist eine ausgeprägte muskuläre Hypotonie sowie eine ausgeprägte Krampfanfallneigung. Die Kinder haben oft massive Atemwegs- und Schluckprobleme. Das Erscheinungsbild ist geprägt durch eine Mikrocephalie mit einer hohen Stirn und hervorstehendem Hinterhaupt. Im Bereich der Schläfen finden sich beidseits Eindellungungen. Das Gesicht weist eine schmale Oberlippe mit einem auffällig langen Philtrum auf. Es finden sich Ohrmuscheldysplasien sowie eine breite Nasenwurzel. Im Gehirn finden sich aufgehobene Furchen und Falten bis hin zum Vollbild einer Lissenzephalie. Die Hirnrinde ist verdickt und es finden sich erweiterte Hirnventrikel. Es kann eine Balkenagenesie oder -hypoplasie vorliegen.Begleitend können je nach Ausmaß der mitbeteiligten Gene eine Mikrogenie, Herzfehler wie z.B. eine Fallot-Tetralogie, Nierenfehlbildungen, Fehlbildungen des Magen-Darmtraktes, ein Kryptorchismus, Korneatrübungen sowie Kampto- und Klinodaktylien auftreten. (nach http://de.wikipedia.org/wiki/Miller-Dieker-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Minimal residual disease (MRD)
| Methode: | Karyotyperstellung / FISH bei Vorliegen eines zytogenetischen Markers |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Myelodysplastisches Syndrom (MDS)
| Methode: | Karyotyperstellung / FISH |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Myeloproliferatives Syndrom (MPS)
| Methode: | Karyotyperstellung / FISH |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
N
Neurofibromatose Typ 1Neurofibromatose Typ 1
| Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) optional: Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA optional für FISH: 2 – 5 ml Heparin-Blut |
| Dauer: |
ca. 8 Wochen |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Die Neurofibromatose Typ 1 (kurz: NF1), auch Morbus Recklinghausen (benannt nach Friedrich Daniel von Recklinghausen) oder Periphere Neurofibromatose, ist eine autosomal-dominant und monogen vererbte Multiorganerkrankung, die vor allem Haut und Nervensystem betrifft. Sie wird daher den neurokutanen Erkrankungen (Phakomatosen) zugeordnet. Typische Veränderungen an der Haut sind mehrere Café-au-lait-Flecken sowie Neurofibrome. Im zentralen Nervensystem (ZNS) treten gehäuft Tumoren verschiedener Lokalisation auf. Patienten können minderbegabt sein und an epileptischen Anfällen leiden. Des Weiteren sind regelmäßig Augen und Knochen mitbetroffen.
Als Café-au-lait-Flecken bezeichnet man milchkaffeefarbene Hyperpigmentierungen der Haut. Sie liegen im Niveau der Haut, können bei allen Menschen auftreten und sind harmlos. Bei Menschen mit einer NF1-Mutation treten sie gehäuft auf.
Als Neurofibrom bezeichnet man gutartige Tumoren, die von den Zellen der Schwann’schen Scheiden, kleiner in der Haut verlaufender Nervenfasern, ausgehen.
Eine Neurofibromatose wird durch eine Veränderung in einem Gen hervorgerufen, welches normalerweise hemmend auf die Zellteilung Einfluss nimmt. Es kommt daher zu überschießender Gewebsvermehrung und damit zu den typischen Veränderungen. Die Diagnose wird meist anhand des klinischen Bildes bereits in der Kindheit gestellt. Da es sich bei Morbus Recklinghausen um eine genetische Erkrankung handelt, ist eine Therapie, welche ihre Ursache beseitigt, aktuell nicht möglich. Es werden daher nur Veränderungen behandelt, die für den Patienten störend oder gefährlich sind.
Eine weitere bekannte Form der Neurofibromatose ist die Neurofibromatose Typ 2 (NF2). (nach http://de.wikipedia.org/wiki/Neurofibromatose_Typ_1)
Ansprechpartner:
Arbeitsgruppenleiterin
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Neurofibromatose Typ 2
| Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) optional: Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA optional für FISH: 2 – 5 ml Heparin-Blut |
| Dauer: |
ca. 8 Wochen |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Die Neurofibromatose Typ II (NF II), auch als zentrale Neurofibromatose bezeichnet, ist eine erbliche Tumorerkrankung. Ihr Hauptmerkmal ist das Vorkommen von gutartigen Hirntumoren, die sich symmetrisch im Bereich beider Hör- und Gleichgewichtsnerven entwickeln. Die meisten Patienten mit dieser Erkrankung leiden auch an Veränderungen der Augen. Ursache der NF II sind Mutationen eines Gens, das vermutlich Einfluss nimmt auf Form und Wanderungsverhalten bestimmter Zelltypen. Da die NF II genetisch bedingt ist, ist eine Heilung nicht möglich. Die Behandlung besteht in der Entfernung von Tumoren im Bereich des Gehirns und Rückenmarkes, sowie operativer Eingriffe im Bereich der Augen und der betroffenen Hirnnerven.
Die NF II ist etwa zehnmal seltener als die häufigste Form der Neurofibromatose, die periphere Neurofibromatose Typ 1 (Morbus Recklinghausen). (nach http://de.wikipedia.org/wiki/Neurofibromatose_Typ_2)
Ansprechpartner:
Arbeitsgruppenleiterin
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Noonan-Syndrom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
O
OrgantransplantationOrgantransplantation
| Methode: | PCR-SSP, Next Generation Sequencing (NGS) |
| Material: |
mindestens 10 ml EDTA- oder Citrat-Röhrchen (je nach Lymphozytengehalt) |
| Dauer: | 2-8 Arbeitstage (je nach Methodik und Probeneingang) |
Anforderungsschein: Herunterladen
Bei Lebendspenden aller Organe und bei Aufnahme auf Wartelisten aller Organe außer Niere führen wir Typisierungen an den Loci HLA-A, -B, -C, -DRB1 und DQB1 von low-resolution SSP bis high resolution NGS durch.
In Ergänzung dazu führen wir auch retrospektive Typisierungen bei Nachsorgeuntersuchungen transplantierter Patienten durch. Dies dient der Abklärung der Donorspezifität von HLA-Antikörpern bei Abstoßungsreaktionen. Bei Bedarf führen wir dazu auch eine Typisierung an zusätzlichen Loci, wie z.B. HLA-DPB1, -DPA1 und DQA1 durch.
Ansprechpartner:

P
PDGFR-Aberrationen bei MPS /MDSPDGFR-Aberrationen bei MPS /MDS
| Methode: | FISH mit PDGFRA - und PDGFRB-spezifischen Sonden |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional: 3 – 4 Ausstriche auf Objektträger |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Peutz-Jeghers-Syndrom
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Plasmozytom / Multiples Myelom
| Methode: | FISH, Nachweis/Ausschluss del(13)(q) und t(4;14) bzw. t(11;14); del(17)(p13)
Karyotyperstellung |
| Material: |
2 - 5 ml Heparin-Blut 5 - 7 ml Heparin-Knochenmark optional für FISH: 3 – 4 Ausstriche auf Objektträger Plasmazellzahl erforderlich (mindestens 10% nötig) |
| Dauer: | ca. 2 - 14 Tage |
Arbeitsgruppenleiterin,
Fachhumangenetikerin der GfH (Tumorgenetik und Zytogenetik)
Prader-Willi/Angelman-Syndrom
| Methoden: |
MLPA (methylierungsspezifisch) optional: PCR, Mikrosatellitenanalyse zur Abklärung einer UPD FISH (Bestätigungsanalyse) |
| Material: |
2 - 5 ml EDTA-Blut bei UPD-Analyse auch EDTA-Blut der Eltern notwendig 2 - 5 ml Heparin-Blut (optional für Bestätigungsanalyse) |
| Dauer: |
ca. 1-2 Wochen |
Arbeitsgruppenleiterin
Ansprechpartner FISH:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Weiterführende Informationen
Pathophysiologie und Genetik:
Das Prader-Willi-Syndrom kommt mit einer Häufigkeit von 1:10.000- 1:15.000 vor und ist im Neugeborenenalter durch eine ausgeprägte Muskelhypotonie (floppy infant) und eine Trinkschwäche gekennzeichnet. Später entwickeln die Kinder meist ein zwanghaftes Hungergefühl, welches in der Folge zu starkem Übergewicht führen kann. Es kann eine Entwicklungsverzögerung auftreten.
Das Angelman-Snydrom tritt mit einer Häufigkeit von 1:15.000 - 1:20.000 auf. Die Kinder sind mental retardiert, neigen zu unkontrollierten Lachanfällen (happy puppet) und zeigen in der Regel keine oder nur eine geringe Sprachentwicklung. Ataxien und Epilepsie kommen gehäuft vor.
Ursächlich für das Prader-Willi-/Angelman-Syndrom ist eine Veränderung in der chromosomalen Region 15q11-13. Diese Region unterliegt dem Imrprinting. Beim Gesunden ist das väterliche Allel unmethyliert (also aktiv), das mütterliche methyliert (also inaktiv). Bei Veränderungen in dem Bereich 15q11-13 kann zu Verschiebungen dieses Methylierungsmusters kommen.
Beim Prader-Willi-Syndrom findet man bei 70 % der Patienten eine Deletion des paternalen Allels, eine maternale UPD 15 (29 %) oder Imprinting-Defekte (1%). Als Folge kann nur die inaktive mütterliche Kopie abgelesen werden.
Beim Angelman-Syndrom liegt eine Deletion des mütterlichen Allels in 50-80 %, eine paternale UPD in 2-5 %, eine UBE3A-Mutation in 8-11 % und ein Imprinting-Defekt in 5 % der Fälle vor. In Folge dessen kann nur die väterliche Kopie abgelesen werden.
Diagnostik:
Genomische DNA, isoliert aus EDTA-Blut, wird zunächst mittels einer MLPA-Analyse (Multiplex Ligation-dependent probe amplification) analysiert (MCR-Holland, ME028B1). Dabei können sowohl Deletionen als auch Methylierungsfehler im Bereich der chromosomalen Region 15q11-12 nachgewiesen werden bzw. ausgeschlossen werden.
Für das Prader-Willi-Syndrom können mit dieser Methode dadurch mehr als 99 % aller Ursachen (paternale Deletion, maternale UPD 15, Imprinting-Defekte) untersucht, jedoch nicht zwischen ihnen unterschieden werden. In jedem Fall wird bei einem auffälligen MLPA-Befund eine Abklärung mittels FISH (bei Verdacht auf Deletion in der PWS-AS-Region) oder UPD-Analyse (bei auffälligem Methylierungsmuster) empfohlen. Bei unauffälligem MLPA-Befund ist ein Prader-Willi-Syndrom sehr unwahrscheinlich
Für das Angelman-Syndrom können die Ursachen maternale Deletion, paternale UPD, Imprinting-Defekte untersucht werden. Mutationen im UBE3A-Gen, die ca. 8-11 % aller Angelman-Fälle bedingen, können mit der Methode nicht nachgewiesen werden. Bei unauffälligem MLPA-Befund und fortbestehendem Verdacht auf Angelman-Syndrom sollte daher eine Mutationsanalyse des UBE3A-Gens durchgeführt werden. Diese Analyse führen wir selbst nicht durch, sondern leiten nach Anforderung das Material gern weiter.
Pränataler Schnelltest
| Methode: | Mikrosatelliten-Multiplex-PCR zur Untersuchung auf numerische Aberrationen der Chromosomen 13, 18, 21, X, Y |
| Material: |
Chorion (Placenta): mind. 10 mg in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) Fruchtwasser: 15 - 20 ml in sterilem Probengefäß zum Ausschluss einer mütterlichen Kontamination bei weiblichem Karyotyp bitte 5 ml EDTA-Blut der Mutter mit einsenden) |
| Dauer: | ca. 1 Tag |
Arbeitsgruppenleiterin,
Fachhumangenetikerin (GfH); European registered Clinical Laboratory Geneticist (ErCLG)
R
RetinoblastonRetinoblaston
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Rubinstein-Taybi-Syndrom
| Methode: | Metaphase-FISH (Sonde für CBP-Gen Exon 14-31) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Rubinstein-Taybi-Syndrom (RTS) ist eine genetisch bedingte Erkrankung, die mit moderater geistiger Behinderung und körperlichen Missbildungen einhergeht. Das Syndrom wurde 1957 zum ersten Mal in einer Studie beschrieben. Die Namensgeber Jack Herbert Rubinstein und Hooshang Taybi beschrieben dieses Syndrom erstmals 1963.
Die allgemeine Häufigkeit (Inzidenz) liegt bei 1:120.000. RTS wird oft erst im Teenageralter diagnostiziert, da Varianten der Ausprägung sehr groß sind. Beim Rubinstein-Taybi-Syndrom zeigen sich Genmutationen des CBP-Gens, in 10–25 % Mikrodeletion auf dem kurzen Arm des Chromosoms 16 [16p13.3) und des p300-Gens auf dem langen Arm des Chromosoms 22 (22q13.2). Sonden stehen nur für 16p13.3 zur Verfügung. (nach http://de.wikipedia.org/wiki/Rubinstein-Taybi-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
S
SchwangerschaftSchwangerschaft
| Methode: | PCR-SSP |
| Material: |
EDTA- oder Citrat-Blutröhrchen |
| Dauer: |
2-4 Arbeitstage |
Anforderungsschein: Herunterladen
Bei immunologischen Problemen während Schwangerschaften und kurz nach der Geburt (z.B. NAIT) führen wir sowohl HPA-Typisierungen, als auch HLA-Typisierungen an ausgesuchten Loci von Vater, Mutter und Kind durch.
Ansprechpartner:
SCN9A-vermittelte Schmerzsyndrome
| Erkrankung | Gensymbol | OMIM-Eintrag |
| Congenital Indifference to Pain | SCN9A | OMIM 243000 |
| Primary Erythermalgia | SCN9A | OMIM 133020 |
| Paroxysmal extreme pain disorder | SCN9A |
|
| Methode: |
Next-Generation-Sequenzierung (NGS) |
| Material: |
500 ng DNA oder 2-5 ml EDTA-Blut |
| Dauer: | ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Ansprechpartner:
Arbeitsgruppenleiterin
Pathophysiologie und Genetik:
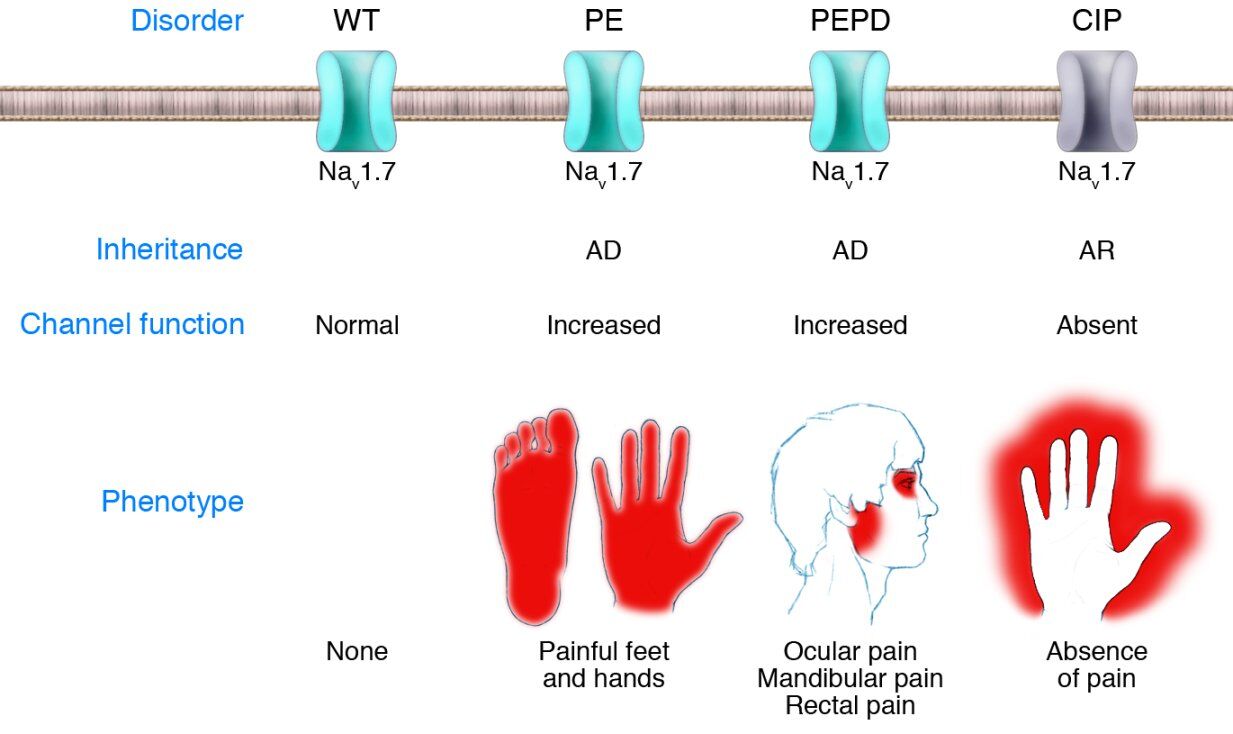
Das SCN9A-Gen kodiert für eine Untereinheit des Natriumkanals Nav1.7. Dieser Ionenkanal spielt eine essentielle Rolle bei der Weiterleitung von Schmerzreizen aus der Körperperipherie. Sog. "Funktionsgewinnmutationen" bei Patienten führen zu einer fehlgesteuerten Schmerzempfindung mit extremen Schmerzzuständen mit den Bezeichnungen "Primary Erythermalgia (PE)" und "Paroxysmal Extreme Pain Disorder (PEPD)". Beide Erkrankungen werden autosomal-dominant vererbt und gehen auf unterschiedliche Mutationen im SCN9A-Gen zurück. Ein von Geburt an vollständiges Fehlen der Schmerzwahrnehmung (Congenital Indifference to Pain, CIP) ist das Merkmal der autosomal-rezessiv vererbten SCN9A-vermittelten Erkrankung. Hierbei kommt es aufgrund von Mutationen im SCN9A-Gen zu einem vollständigen Funktionsverlust. Differentialdiagnostisch ist bei der zuletzt genannten Erkrankung auch an die Hereditären Sensorisch-Autonomen Neuropathien (HSAN) zu denken.
Übersicht SCN9A-vermittelter Schmerzsyndrome:
aus: Drenth JP, Waxman SG., J Clin Invest. 2007 Dec;117(12):3603-9
Short Stature critical Region Xp22
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Bei Kleinwuchs unklarer Genese kann eine Mikrodeletion in der Region Xp22 die Ursache sein. In solchen Fällen kann ein Test zum Ausschluss eines solchen Umbaus sinnvoll sein.
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Smith-Magenis-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: |
5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Smith-Magenis-Syndrom (SMS) wurde in den frühen 1980er Jahren von den Genetikerinnen Ann Smith und Ellen Magenis entdeckt. Bei SMS handelt es sich um einen Symptomenkomplex, der aus einem Gendefekt (meist Mikrodeletion) am kurzen Arm eines Chromosoms 17 herrührt. Es sind Fallbeispiele bekannt, bei denen Personen mit Smith-Magenis-Syndrom keine Deletion, sondern nur eine Mutation am RAI1-Gen aufweisen. Diese Veränderungen am RAI1-Gen bzw. dessen Verlust führt wahrscheinlich zu einer Produktion eines nicht funktionierenden RAI1-Proteins. Die Bandbreite der möglichen Erscheinungen reicht von geistiger Entwicklungsverzögerung und körperlichen Auffälligkeiten ohne nennenswerte funktionelle Beeinträchtigungen bis hin zu geistiger Behinderung, schwer beherrschbaren Verhaltensstörungen und ernsten organischen Erkrankungen. (nach http://de.wikipedia.org/wiki/Smith-Magenis-Syndrom und www.smith-magenis.de/)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Sotos Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Sotos-Syndrom wurde zum ersten Mal 1964 von Juan F. Sotos beschrieben. Am Beispiel von fünf Kindern stellte er Symptome wie ein von Geburt an beschleunigtes Körperwachstum, ein dem Lebensalter gegenüber fortgeschrittenes Knochenalter, einen Makrocephalus (überproportional großer Schädelumfang) sowie eine deutliche Verlangsamung der motorischen, kognitiven und sprachlichen Entwicklung dar. Seitdem wurden mehr als 200 Fälle in der medizinischen Literatur beschrieben. Teilweise wird das Sotos-Syndrom auch als "cerebraler Gigantismus" bezeichnet.
Bei ca. 2/3 aller Patienten mit Sotos-Syndrom finden sich Veränderungen im NSD1-Gen (nuclear receptor binding, enhancer of zeste and trithorax domain protein) auf Chromosom 5 (5q35). Bei einer geringen Anzahl europäischer Sotos-Patienten wurde eine Deletion, bei der Mehrheit aller Sotos-Patienten wurde eine Punktmutation im NSD1-Gen gefunden. Die Erkrankung ist autosomal dominant erblich. Punktmutationen bei Betroffenen treten in der Regel de novo auf. (nach http://de.wikipedia.org/wiki/Sotos-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
SRY
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Sex determining region of Y-Gen (SRY), kurz als SRY-Gen bekannt, codiert einen Transkriptionsfaktor – den Hoden-determinierenden Faktor (TDF für engl. Testis-determining factor) –, der zur (HMG)-Box-Proteinfamilie von DNA-Bindungsproteinen gehört. Das SRY-Gen wird neben anderen Genen zur Geschlechtsdetermination beim Menschen verwendet. SRY liegt im Normalfall auf dem kurzen Arm des Y-Chromosom des Menschen. Entsprechend haben Menschen, die dieses Chromosom mit dem entsprechenden Gen besitzen, im Normalfall einen männlichen Phänotyp. Dabei ist es unerheblich, wie viele Kopien des X-Chromosoms vorliegen, auch Menschen mit einem multiplen X-Chromosom (Klinefelter-Syndrom) haben diesen. Das durch das Gen codierte Protein TDF steuert die weitere Entwicklung zum männlichen Geschlecht. (nach http://de.wikipedia.org/wiki/Sex_determining_region_of_Y)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Stammzelltransplantationen
| Methode: | Next Generation Sequencing (NGS), Sequenzierung nach Sanger, PCR-SSP |
| Material: |
mindestens 10 ml EDTA- oder Citrat-Röhrchen (je nach Lymphozytengehalt) |
| Dauer: |
2-8 Arbeitstage (je nach Methodik und Probeneingang) |
Anforderungsschein: Herunterladen
Untersuchung des Patienten/Empfängers:
Patienten werden zweimal mit NGS (Next Generation Sequencing) an den Loci HLA-A, -B, -C, -DRB1 und DQB1 typisiert.
Familientypisierung:
Bei der Familientypisierung werden zwei Situationen unterschieden:
Ist Material beider Elternteile vorhanden werden die Familienangehörigen des Patienten vorerst niedrig auflösend typisiert und die Haplotypen der Familie bestimmt. Dadurch sind wir dann in der Lage die Kompatibilität der Geschwister mit dem Patienten zu bestimmen. Kommt ein Geschwister als Spender in Frage wird aus der vorhandenen DNA eine hochauflösende Typisierung nachgeholt.
Sind die Eltern nicht typisierbar werden die Familienangehörigen des Patienten sofort hochauflösend durch Sequenzierung (NGS, Sanger) typisiert. Eine Bestimmung der Haplotypen ist dann nicht notwendig. Das Ergebnis der hochauflösenden Typisierungen wird mit dem Patienten abgeglichen und die Kompatibilität bestimmt. In beiden Fällen erfolgt vor einer Transplantation eine niedrig auflösende SSP-Typisierung des Familienspenders als Bestätigungstypisierung.
Fremdspender-Bestätigungstypisierung (CT):
Die HLA-Typisierung von anonymen Fremdspendern werden vor der Stammzelltransplantation hochauflösend mit Sequenzierung (NGS, Sanger) überprüft, um die Identität und die Vortypisierung der Fremdspenderdatei abzusichern und den Matchgrad mit dem Patienten zu bestimmen.
Ansprechpartner:
Steroidsulfatase-Defizienz /X-gebundene Ichthyosis
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Bei der X-chromosomal-rezessiven Ichthyosis vulgaris handelt es sich um eine genetisch bedingte Verhornungsstörung der Haut, die sich in den ersten sechs Lebensmonaten mit großen polygonalen, gelbbraunen bis hellgrauen Hyperkeratosen mit breiten hautfarbenen Spalten manifestiert. Besonders ausgeprägt betroffen sind die Kopfhaut, die Ohren und der Nacken, während das Gesicht, die Handflächen und Fußsohlen sowie die großen Körperbeugefalten ausgespart bleiben. Die Handflächen und Fußsohlen zeigen keine vertiefte und vermehrte Linienzeichnung. In 50 % der Fälle findet man eine nicht beeinträchtigende Hornhautbeteiligung. Ferner besteht bei etwa jedem fünften Betroffenen ein Hodenhochstand (Kryptorchismus). Diese Ichthyoseform ist die zweithäufigste aus dem Formenkreis der Ichthyosen mit einer geschätzten Häufigkeit in der Bevölkerung von 1:4000. Das Vollbild der Erkrankung findet sich nur beim männlichen Geschlecht. Weibliche Überträgerinnen können in einigen Fällen charakteristische Hornhautveränderungen und gelegentlich eine geringe Schuppung der Haut besonders an den Unterschenkeln aufweisen.
Diese Ichthyoseform wird X-chromosomal rezessiv vererbt und durch eine Mutation im STS-Gen (Steroidsulfatase-Gen), welches auf dem kurzen Arm des X-Chromosoms (Xp22.32) liegt, verursacht. Die Genmutation führt zu einem Enzymmangel (Steroidsulfatase-Defizienz), der durch mangelnde Abschilferung der Hautzellen zu einer übermäßigen Verhornung führt (Retentionshyperkeratose). Mehr als 90% der Patienten zeigen eine komplette Deletion des STS-Gens und flankierender Sequenzen; nur bei einer Minderheit findet man Punktmutationen oder partielle Deletionen. (nach http://www.uke.de/institute/humangenetik/index_46657.php)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
T
Thorakale Aortenerweiterung/ Aortendissektion/ AortenaneurysmaThorakale Aortenerweiterung/ Aortendissektion/ Aortenaneurysma
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Weiterführende Informationen
Ansprechpartner:
Arbeitsgruppenleiterin
Transfusion kompatibler Blutprodukte
| Methode: | PCR-SSP |
| Material: |
mindestens 10 ml EDTA- oder Citrat-Röhrchen (je nach Lymphozytengehalt) |
| Dauer: | 2-4 Arbeitstage Je nach Probeneingang |
Anforderungsschein: Herunterladen
Zur Abklärung der Gabe von kompatiblen Blutprodukten z.b. nach refraktärer Gabe von Plättchenkonzentraten, typisieren wir die Loci HLA-A, HLA-B und HLA-C low resolution mittels PCR-SSP.
Ansprechpartner:
Trisomie 21 Verdacht
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Down-Syndrom ist ein Syndrom beim Menschen, bei dem durch eine Genommutation (Chromosomenaberration/ Polyploidie) das gesamte Chromosom 21 oder Teile davon dreifach vorliegen (Trisomie). Daher lautet eine weitere übliche Bezeichnung Trisomie 21. Menschen mit Down-Syndrom weisen in der Regel typische körperliche Merkmale auf und sind in ihren kognitiven Fähigkeiten meist beeinträchtigt.
Die verschiedenen Formen der Trisomie 21 entstehen spontan und können nur vererbt werden, wenn die Mutter bereits selbst das Down-Syndrom hat. Eine Form des Down-Syndroms kann allerdings familiär gehäuft vorkommen, sofern eine balancierte Translokation eines 21. Chromosoms bei einem Elternteil ohne Down-Syndrom vorliegt. Dies begünstigt das Auftreten der Translokations-Trisomie 21 beim Kind. Daher ist in manchen Fällen eine FISH Untersuchung auf kryptische Umbauten unter Beteiligung von Chromosom-21-Material indiziert. (nach http://de.wikipedia.org/wiki/Down-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Ü
Überprüfung von ZellmosaikenÜberprüfung von Zellmosaiken
| Methode: | Fluoreszenz-in-situ-Hybridisierung an Interphasekernen |
| Material: |
Mundschleimhautabstrich, Spatelabstrich (in sterilem Medium oder Kochsalzlösung) |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Der Begriff „genetischer Mosaizismus“ bezeichnet die gleichzeitige Präsenz von zwei oder mehr Zellpopulationen mit unterschiedlichen Genotypen in einem Individuum. Mosaike sind, entgegen der landläufigen Annahme, dass sie nur ausnahmsweise auftreten, ein gängiges und häufig vorkommendes Phänomen; es seien im Folgenden nur vier Bespiele hierfür genannt:
- Jeder Mensch besteht z.B. aus Zellen mit Zellkernen (= die überwiegende Mehrheit), und Zellen ohne Zellkerne (z.B. die roten Blutkörperchen, oder die Zellen der Pupille);
- aufgrund der X-Inaktivierung stellt jeder weibliche Säugetierkörper ein komplexes Zellmosaik dar, da in einem Teil der Zellen das paternale X-Chromosom, und im anderen Teil das maternale X-Chromosom inaktiviert wurde;
- in den Zellen des Immunsystems werden unterschiedliche genomische Bereiche miteinander rekombiniert – genetisch unterscheiden sich hier auch eineiige Zwillinge voneinander; oder
- somatischer Moasizismus wurde im Zusammenhang mit Alterungsprozessen gefunden.
In der genetischen und zytogenetischen Routinediagnostik können die o.g. Aspekte jedoch normalerweise beiseite gelassen werden, weshalb ein (ausgeprägter) Mosaikbefund für Diagnostiker und genetische Berater oftmals überraschend kommt. Der Nachweis eines Mosaiks ist in der Regel am einfachsten und eindeutigsten auf Einzelzellniveau zu treffen, d.h. mittels Zytogenetik oder Molekularer Zytogenetik. Einschränkend ist anzumerken, dass hierfür die nachgewiesenen genetischen Veränderungen grobstruktureller oder numerischer Art sein müssen um (molekular)zytogenetisch erfassbar zu sein.
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
U
Uniparentale DisomienUniparentale Disomien
UPD6, UDP7, UPD11, UPD13, UPD14, UPD15, UPD18, UPD21
(andere UPDs nur nach vorheriger telefonischer Rücksprache)
| Methode: | PCR, Mikrosatellitenanalyse |
| Material: |
2 - 5 ml EDTA-Blut pränatal: Chorion (Placenta): mind. 10 mg in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) Fruchtwasser: 15 - 20 ml in sterilem Probengefäß
Zur Untersuchung wird EDTA-Blut oder DNA von Mutter, Vater und Kind/Fetus benötigt. Da die Untersuchung auch Aufschluss über die Vaterschaft gibt, sollte dies im Sinne einer Einverständniserklärung mit der Familie diskutiert werden. |
| Dauer: | ca. 1-2 Wochen |
Ansprechpartner:
Arbeitsgruppenleiterin
Weiterführende Informationen
Die uniparentale Disomie (UPD) wird durch das Vorhandensein der beiden Homologen eines Chromosomenpaares von nur einem Elternteil definiert. Entweder finden sich dabei die beiden homologen (Heterodisomie) oder zwei Kopien eines homologen Chromosoms (Isodisomie), bzw. eine Mischung von heterodisomen und isodisomen Segmenten.
Eine UPD kann verschiedene Ursachen haben. Die beiden wichtigsten sind:
- Trisomy Rescue: Verlust des singulären Homologen bei einer trisomen Zygote.
- Monosomy Rescue: Somatische Reduplikation des in der Zygote monosom vorliegenden Chromosoms.
Es kann in der Folge zur Entstehung von Iso- bzw. Heterodisomien kommen.
Mit einem uniparental vorliegenden Chromosomenpaar können folgende Probleme assoziiert sein:
- Mosaik Trisomie
- Homozygotisierung von autosomal rezessiv vererbten Mutationen
- Unphysiologische genomische Prägung (Genomic Imprinting)
Hintergrund
Nach bisherigem Wissen beeinflussen uniparentale Disomien von nur wenigen Chromosomen den Phänotyp.
Im Einzelnen sind dies:
- upd(6)pat: Transienter neonataler Diabetes mellitus
- upd(7)mat: Russell-Silver Syndrom*
- upd(11)pat: Beckwith-Wiedemann Syndrom (BWS)*
- upd(13)pat or mat: kein Imprinting bekannt; nur iso-UPD ggf. relevant
- upd(14)mat: Kleinwuchs, Hypotonie, überstreckbare Gelenke, Skoliose, geringe faciale Dysmorphien, milde Entwicklungsretardierung, frühzeitige Pubertät
- upd(14)pat: stärkere Ausprägung als bei upd(14)mat sowie zusätzlich mentale Retardierung und skeletale Abnormalitäten, die zu einen Kleinwuchs führen
- upd(15)mat: Prader-Willi Syndrom (PWS)*
- upd(15)pat: Angelman Syndrom (AS)*
- upd(18)pat or mat: kein Imprinting bekannt; nur iso-UPD ggf. relevant
- upd(21)pat or mat: kein Imprinting bekannt; nur iso-UPD ggf. relevant
- Weitere bisher nur mit geringen Fallzahlen belegte UPDs sind im Review von Kotzot und Uttermann, AJMG, 2005 zusammengefaßt
* eine UPD ist nur bei ca. 20% der BWS-, bei ca. 28% der PWS- und bei ca. 3-5% der AS-Fälle nachweisbar. Beim Russel-Silver Syndrom wurden auch chromosomale Veränderungen nachgewiesen. Eine weiterführende Diagnostik muss mit anderen Methoden durchgeführt werden und kann nach Rücksprache vermittelt werden.
Generell kann eine UPD-Diagnostik sinnvoll sein bei:
- Feten mit einer Robertsonschen Translokation oder einem Isochromosom für die Chromosomen 14 und 15
- Kindern mit einer Robertsonschen Translokation oder einem Isochromosom für die Chromosomen 14 oder 15 und Wachstumsverzögerung und/oder mentaler Retardierung
- Feten mit chromosomalen Markern, die als Chromosom 6, 7, 11, 14 oder 15 identifiziert wurden
- Auffälligkeiten im Ultraschall, die mit den Merkmalen bei UPD-Syndromen übereinstimmen
- Feten mit einer Mosaik-Trisomie
- Neugeborene mit einem neonatalen Diabetes mellitus
- Neugeborene mit Merkmalen eines Silver-Russell Syndroms
- Neugeborene mit Verdacht auf BWS mit normalen Karyotyp und keiner über FISH nachgewiesenen Duplikation von 11p15.5
Beim Vorliegen anderer Trisomien oder Markerchromosomen ist eine Abklärung ggf. vor dem Hintergrund einer wissenschaftlichen Fragestellung interessant. Dies gilt vor allem für die Chromosomen, für die noch keine UPD (s. o.) beschrieben wurde.
Weiterführende Links
- sSMC (small supernumerary marker chromosome)
Weiterführende Literatur
- Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. American College of Medical Genetics Statement on Diagnostic Testing for Uniparental Disomy. Genetics in Medicine 2001;3:206-211
- Ledbetter DH, Engel E. Uniparental disomy in humans: development of an imprinting map and its implications for prenatal diagnosis. Hum Mol Genet 1995; 4:1757-1764
- Liehr T. Cytogenetic contribution to uniparental disomy (UPD). Mol Cytogenet 2010, 3:8.
- Liehr T. 2014. Uniparental Disomy (UPD) in Clinical Genetics. A Guide for Clinicians and Patients. Springer. ISBN 978-3642552878
- Kotzot D. Abnormal phenotypes in uniparental disomy (UPD): fundamental aspects and a critical review with bibliography of UPD other than 15. Am J Med Genet 1999; 82:265-274
- Kotzot D. Complex and segmental uniparental disomy (UPD) - review and lessons from rare chromosomal complements. J Med Genet 2001; 38:497-507
- Kotzot D, Utermann G. Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A. 2005 Jul 30;136(3):287-305.
W
Williams-Beuren-SyndromWilliams-Beuren-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das Williams-Beuren-Syndrom (WBS), auch bekannt unter den Synonymen Williams-Syndrom, Fanconi-Schlesinger-Syndrom, idiopathische Hyperkalzämie oder Elfin-face-Syndrom, ist eine genetisch bedingte Besonderheit, deren Ursache in einem Stückverlust (= Deletion) auf dem Chromosom 7 liegt. Das Syndrom wurde nach den Kardiologen J. C. P. Williams und dem Alois J. Beuren benannt. Typisch sind Gesichtdysmorphien, kognitive Beeinträchtigungen und oft sprachliche Gewandtheit u.a.m.. Seit 1993 ist bekannt, dass die Ursache des WBS eine Veränderung von mindestens 20 Genen auf dem langen Arm des Chromosoms 7 ist. Dies entsteht meist durch spontane Mutation und wirkt sich dahingehend aus, dass es unter anderem Störungen des Elastin-Gens hervorruft, das für die Bildung von Bindegewebe mitverantwortlich ist. (nach http://de.wikipedia.org/wiki/Williams-Beuren-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
Wolf-Hirschhorn-Syndrom
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
HINWEIS - UNSERE EINRICHTUNG BIETET MOLEKULAR-ZYTOGENETISCHE DIAGNOSTIK DES WHS AN.
WIR SIND NICHT AUF DIESE ERKRANKUNG SPEZIALISIERT.
VON ANFRRAGEN ZUR BEHANDLUNG BITTEN WIR ABZUSEHEN.
Weiterführende Informationen
Das Wolf-Hirschhorn-Syndrom (auch als Wolf-Syndrom oder Chromosom-4p-Syndrom bekannt) ist eine seltene angeborene Erbkrankheit, die durch eine sogenannte strukturelle Chromosomenaberration am kurzen Arm des Chromosom 4 bedingt ist. Leitsymptom ist ein Kleinwuchs verbunden mit einer extremen Verzögerung der geistigen und körperlichen Entwicklung sowie eine Kombination unterschiedlicher Fehlbildungen. Es ist nach Ulrich Wolf und Kurt Hirschhorn benannt, die das Krankheitsbild 1965 unabhängig voneinander erstmals beschrieben.
Ursache ist der Verlust eines kleinen Abschnitts (Deletion) am Ende des kurzen Arms von Chromosom 4. Etwa 85-87 % aller Deletionen entstehen im betroffenen Individuum neu (de novo). Bei De-novo-Deletionen besteht kein erhöhtes Wiederholungsrisiko für weitere Nachkommen der Eltern. EinTeil der Wolf-Hirschhorn-Syndrome wird durch eine sogenannte balancierte Translokationen bei einem Elternteil verursacht. Typisch sind beim charakteristische Gesichtsfehlbildungen mit vergrößertem Augenabstand (Hypertelorismus), nach unten abfallenden Lidachsen, einer breiten Nase, einem verkürzten Philtrum, kleinem Kiefer, nach unten stehenden Mundwinkeln und Ohranhängseln oder -grübchen. Lippen- oder Gaumenspalten werden allgemein als typisch für das Wolf-Hirschhorn-Syndrom beschrieben, treten aber nur bei größeren Deletionen über 9 Mbp auf. Typischerweise sind die Kinder schon bei Geburt untergewichtig und haben einen zu kleinen Kopf. Diese Wachstumsverzögerung setzt sich nach der Geburt fort, das Ausmaß scheint mit dem Ausmaß des Verlustes an Erbsubstanz zu korrelieren. Auch die geistige Entwicklung ist bei allen Kindern verzögert. Nur etwa die Hälfte lernt frei zu sitzen und maximal ein Drittel zu laufen. Zumindest einige Worte sprechen lernt nur etwa jedes fünfte Kind, wobei die übrigen auf eine nonverbale Art kommunizieren können. Etwa 85 % der betroffenen Kinder bekommen eine teilweise schwer zu behandelnde Epilepsie. Zusätzlich können verschiedene Organfehlbildungen auftreten, die vor allem die Augen (Spaltbildung der Regenbogenhaut – Kolobom, Schielen), das Herz, die Nieren und das Skelettsystem in Form von Wirbelsäulenverkrümmung oder Klumpfüßen betreffen. Bei Jungen sind auch Fehlbildungen des Genitales (Hypospadie) beschrieben. (nach http://de.wikipedia.org/wiki/Wolf-Hirschhorn-Syndrom)
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia
X
XIST - X-InaktiverungszentrumXIST - X-Inaktiverungszentrum
| Methode: | Metaphase-FISH (kommerziell erhältliche Sonde) |
| Material: |
2 - 5 ml Heparin-Blut 15 - 20 ml Fruchtwasser in sterilem Probengefäß 10 mg Chorionzotten in sterilem Probengefäß (0,9 % NaCl-Lsg. oder Kulturmedium) alternativ: fixierte Zellen auf 2 -3 Objektträgern |
| Dauer: | 5 - 10 Tage (pränatal); bis zu 6 Wochen (postnatal) |
Anforderungsschein: Herunterladen
Weiterführende Informationen
Das X-Chromosom trägt ein sog. X-Inaktiverungszentrum (XIST-Region). Bei Frauen bewirkt dieses, das eines der beiden X-Chromosomen zu 85% inaktierviert wird. Liegt ein verändertes X-Chromosom vor macht es Sinn zu klären ob es die XIST Region trägt oder nicht. In vielen Fällen wird ein verändertes X-Chromosom mit XIST bevorzugt inaktiviert und es sind weniger klinische Auswirkungen zu erwarten als in Fällen wo XIST auf dem fraglichen X-Chromosom nicht vorliegt.
Ansprechpartner:
Arbeitsgruppenleiter,
Fachhumangenetiker (GfH); European registered Clinical Laboratory Geneticist (ErCLG); Invited Professor of the Yerevan State University, Armenia // Visiting Professor of University of Belgrade, School of Medicine, Serbia